Reference: Silva, P.J. "Assessing the reliability of sequence similarities detected through hydrophobic sequence analysis", Proteins: Structure, Function and Bioinformatics, 70, 1588-1594
|
Author: Pedro J. Silva
Reference: Silva, P.J. "Assessing the reliability of sequence similarities detected through hydrophobic sequence analysis", Proteins: Structure, Function and Bioinformatics, 70, 1588-1594 |
|
Downloads and Supporting Information |
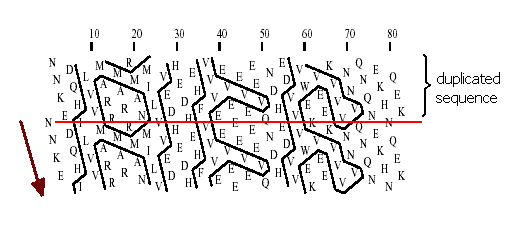
Gaboriaud et al. have shown that sequences with very similar distribution patterns of the hydrophobic set of residues V, I, L, F, Y, W, M (detected in a two-dimensional helical representation of the protein sequence) are most often structural homologs, even when the overall sequence identity is as low as 7 %. This representation is obtained by writing the protein sequence on a classical alpha-helix (3.6 amino acids per turn) smoothed on a cylinder. After five turns, residues i and i+ 18 have similar positions parallel to the axis of the cylinder.To make this 3D representation easier to handle, the cylinder is then cut parallel to its axis and unrolled. As some adjacent amino acids are widely separated by the unfolding of the cylinder, the representation is duplicated, making the sequence easier to follow and giving a better impression of the environment of each aminoacid.
| © 2007 Pedro J. Silva |