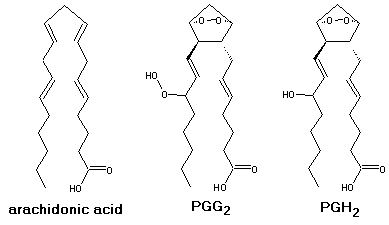
The reaction mechanism of cyclooxygenation of arachidonic acid by prostaglandin H synthase
This page is an abridged version of
Pedro J. Silva, Pedro A. Fernandes and Maria J. Ramos (2003) A theoretical study of radical-only and combined radical/carbocationic mechanisms of arachidonic acid cyclooxygenation by prostaglandin H synthase. Theoretical Chemistry Accounts, 110, 345-351.
Prostaglandin H synthase (also known as cyclooxygenase, or COX )successively incorporates 2 O2 molecules in arachidonic acid, yielding PGH2. It then reduces it to the corresponding alcohol.
|
Prostaglandin H synthase contains two active sites: a
|

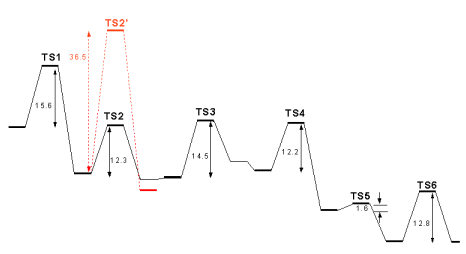
The initial mechanistic proposal for the cyclooxygenase reaction by Hamberg and Samuelsson[1] is shown in the figure above (full arrows). In this mechanism, a protein-based radical abstracts the 13-pro-S hydrogen from bound arachidonic acid, yielding a C11-C15 delocalized pentadienyl radical. The radical sequentially reacts with two molecules of O2, yielding the bicyclic hydroperoxide, PGG2. A number of important findings support the basic tenets of the overall mechanism: the identification of a tyrosyl radical as the initiator of catalysis [2], the determination of X-ray structures of arachidonic acid bound at active site of the protein [3], and the detection of a substrate-based pentadienyl radical by EPR spectroscopy [4]. Dean and Dean [5] suggested that the exquisite stereospecificity of the cyclooxygenase reaction, wich introduces five chiral centers in an achiral molecule, could be explained by the formation of a carbocation center at C10. This would be produced after a sigmatropic H-shift from C10 to the radical present in C13, which yields a C8-C12 delocalized pentadienyl radical (see Figure above, dashed arrows). Loss of an electron from this radical yields a carbocation that may cyclize under orbital simmetry control necessarily yielding a product with the required stereochemistry. After cyclization, the carbocation would be again reduced to the radical and the reaction would proceed as in the radical-only pathway. The observed pentadienyl radical has been shown to encompass the C11-C15 carbons instead of C8-C12 [6], which does not support the combined radical/carbocation mechanism. However, the observed radical might be an artifact ,i.e. thermodynamically but not kinetically favored. It is therefore possible that cyclooxygenation proceeds more readily through the radical/carbocation mechanism than through the radical-only mechanism. We have performed a density-functioanl (DFT B3LYP/6-311+G(3df,2p)//B3LYP/6-31G*) study on both mechanisms, thus elucidating on the correct one.
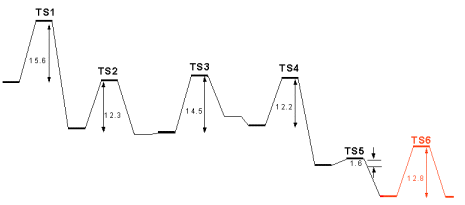
| According to both proposals, the first step in the cycloxygenase mechanism should be the abstraction of the 13-pro-S hydrogen by Tyr385, as depicted in Figure at the left, where (Z,Z)-2,5-heptadiene models the C10-C16 moiety of the substrate and a phenoxy radical models the tyrosyl sidechain. At the transition state, the abstracted hydrogen lies at almost equal distances from carbon C13 and Tyr385 O. The spin density, which in the reactants was fully localized in Tyr385, is now more evenly distributed between the fatty acid substrate (0.54) and the tyrosine residue (0.42). Almost no spin density is present in the transferred 13-pro-S hydrogen, showing that the process is a coupled proton-electron transfer, rather than a pure H atom transfer. In the products, the spin is fully delocalized across the substrate C11-C15 atoms. The activation Gibbs free energy of this reaction is 15.6 kcal.mol-1, in excellent agreement with the experimentally derived value of 15.0 kcal.mol-1 (calculated from the data in [7]). |
| According to the radical-only mechanism, after the first step oxygen attack occurs at the C11 atom, yielding a peroxy radical. The reaction is depicted at the right. In the transition state, the O2 molecule is located at 2.11 Å from C11 and large spin densities are present both at the inner and outer oxygen atoms (0.69 and 0.72, respectively), with smaller spins at the C12-C15 carbon atoms. As the C-O distance finally reduces to 1.48 Å in the products, the spin becomes mostly localized on the outer oxygen atom . The activation Gibbs free energy of this step is 12.3 kcal.mol-1 and the reaction is essentially reversible, with an overall decrease of free energy of -1.4 kcal.mol-1. |
| A 69º clockwise rotation of the peroxy radical along the C-O bond axis brings the radical-containing outer oxygen atom in the correct position for the attack on C9. This rotation increases the reagents’ free energies by only 0.3 kcal.mol-1. In the transition state, the outer oxygen atom is located at 1.90 Å from C9 and the electron spin is already quite delocalized back into the carbon chain (0.68 in C8). The activation Gibbs free energy of this step is 14.5 kcal.mol-1 and the reaction is slightly unfavourable thermodinamically, with an increase of free energy of 4.2 kcal.mol-1. |
| The C8-based radical formed in the previous step then reacts with C12, thereby introducing a new C-C bond in the molecule. In the transition state the C8-C12 distance has reduced from the initial value of 4.20 Å to 2.38 Å, still 50% longer than the final C-C bond distance. As the reaction moves to completion, electron spin eventually delocalizes to the C13-C15 portion of the substrate (0.70 in C13, -0.30 in C14 and 0.66 in C15). The activation free energy of this step is 12.2 kcal.mol-1 and the reaction is quite favourable with a ΔG of -10.1 kcal.mol-1. |
| Addition of an oxygen molecule to the si face of C15 yields the peroxy radical of PGG2. The activation free energy of this step is very small, 1.6 kcal.mol-1, arising mostly from the loss of entropy due to the decrease of O2 translational and rotational degrees of freedom. Indeed, the activation Δ>H is actually negative (-8.3 kcal.mol-1), showing that the reaction is mostly diffusion-controled. The reaction is spontaneous with a ΔG of -8.0 kcal.mol-1. The large energy differences between the addition of the two O2 molecules are not surprising, and are due to the smaller reactivity of the more delocalized radical present in step 2. |
| The final step in the mechanism is hydrogen atom abstraction from Tyr385 by the peroxy radical, which regenerates the tyrosyl radical for a new round of catalysis. We have used a truncated model for the prostaglandin substrate, including only its C12-C16 moiety, since electronic effects from the cyclopentane ring should not affect the chemistry at the outer peroxydic oxygen in C15. As in step 1 above, a phenoxy radical models the tyrosyl sidechain. At the transition state, the abstracted hydrogen lies at almost equal distances from Tyr385 O and the outer peroxydic oxygen (Figure 6). The electron spin, which in the reactants was largely located at this oxygen, has now migrated largely to the tyrosine residue. As in the first hydrogen transfer, above, almost no spin is present in the transferred hydrogen, showing that this process is also a coupled proton-electron transfer, rather than a pure H atom abstraction. In the products, the spin is fully delocalized in the tyrosine residue. The activation free energy is 12.8 kcal.mol-1 and the reaction has a ΔG of -0.2 kcal.mol-1. |
| In the radical/carbocation mechanism, O2 addition to C11 is preceded by a series of events triggered by H-transfer from C10 to the C13-based radical. The transition state of this reaction is shown in Figure 7. The spin distribution in this structure is almost symmetrical, as are the C10-H and C13-H distances. As in the other reactions described, the transferred H-atom carries almost no spin. Still, its 0.08 spin is the largest of those found on transferred H atoms in this study. The activation free energy of this reaction 36.5 kcal.mol-1, more than 20 kcal.mol-1 higher than the experimental barrier, which effectively discards this mechanism for the cyclooxygenase reaction.
|
[1] Hamberg, M.; Samuelsson, B. J. Biol. Chem. 242, (1967), 5336-5343
[2] Goodwin, D. C.; Gunther, M. R.; Hsi, L. C.; Crews, B. C.; Eling, T. E.; Mason, R. P.; Marnett, L. J. J. Biol. Chem., 273 (1998),8903
[3] Malkowski, M. G.; Ginell, S. L.; Smith, W. L.; Garavito, R. M. Science 289 (2000), 1933
[4] Tsai, A.-L.; Palmer, G.; Xiao, G.; Swinney, D. C.; Kulmacz, R. J. J. Biol.Chem. 273 (1998) 3888
[5] Dean, A. M.; Dean, F. M. Protein Sci., 8, (1999), 1087
[6] Peng, S.; Okeley, N. M.; Tsal, A.-L.; Wu, G.; Kulmacz, R. J.; van der Donk, W. A. J. Am. Chem. Soc., 124 (2002), 10785
[7] Kulmacz, R.J.; Pendelton, R. B.; Lands, W. E. M. J. Biol. Chem., 269 (1994), 5527
[8] Thuresson, E. D.; Lakkides, K. M.; Rieke, C. J.; Sun, Y.; Wingerd, B. A.; Micielli, R.; Mulichak, A. M.; Malkowski, M. G.; Garavito, R. M.; Smith, W. L. J. Biol. Chem., 276 (2001) 10347