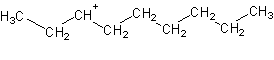
Professor Associado, Universidade Fernando Pessoa
| Outras páginas de Química Orgânica: | |||
| Bioquímica metabólica: |
A quase totalidade dos compostos orgânicos são derivados da petroquímica. Cerca de 90% do petróleo é usado como combustível, e o restante dá origem aos plásticos, às colas, aos detergentes, etc., etc.
Após uma purificação prévia, em que se removem areias e outros detritos, o petróleo é submetido a destilação fraccionada, para se obter os seus diversos componentes:
Após destilação do resíduo sob vácuo, obtêm-se os óleos lubrificantes e os asfaltos.
Para fazer gasolinas a partir de querosene i.e. compostos de cadeia curta a partir de compostos de cadeia mais longa, faz-se o “cracking catalítico” num reactor de leito fluidizado (catalizador Al2O3-SiO2). Neste reactor também existe H2, para reduzir as olefinas (que formariam gomas), o enxofre (que envenena os catalisadores) e o azoto (pois os óxidos de azoto são poluentes). O catalizador removerá hidreto dos hidrocarbonetos, dando origem a intermediários extremamente reactivos: os carbocatiões.
Os carbocatiões têm muita tendência a receber um par de electrões: são electrófilos (ou ácidos de Lewis). O electrões das ligações p (presentes em compostos insaturados) são bons candidatos para reagir com os carbocatiões:
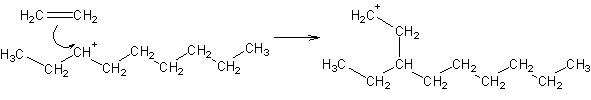
Os carbocatiões também podem sofrer cisões b:
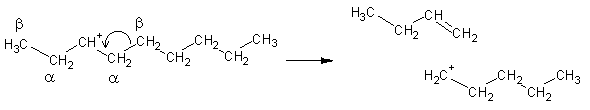
Também podem sofrer rearranjos do esqueleto carbonado:
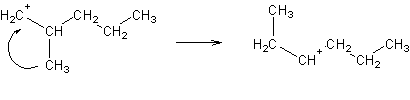
Por razões que se estudarão mais adiante, os carbocatiões terciários (i.e. ligados a 3 átomos de carbono) serão mais estabilizados do que os carbocatiões secundários, e estes mais do que os primários.
Os carbocatiões, principalmente devido a poderem sofrer cisões b, permitirão a síntese de hidrocarbonetos de cadeia curta (gasolinas) a partir de hidrocarbonetos de cadeia mais longa.
Além das gasolinas, um produto muito importante da petroquímica é o etileno. Este é produzido a partir de misturas razoavelmente ricas em etano através de um processo denominado cracking térmico:
![]()
A altas temperaturas o etano sofre cisão homolítica , i.e. a ligação C-C quebra de forma a que os dois electrões da ligação ficam separados: um em cada produto da quebra. Estas reacções denominam-se reações de iniciação.
Os produtos da quebra contêm electrões desemparelhados: são radicais, e altamente reactivos. Estes radicais terão tendência a retirar átomos de hidrogénio de outras moléculas de hidrocarbonetos, dando origem a radicais em cadeias longas. Os radicais actuarão em certos aspectos de forma semelhante aos carbocatiões: por exemplo, também provocam cisões b:
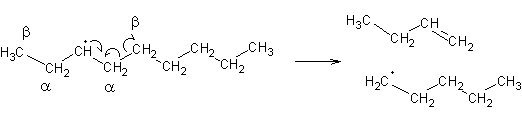
O novo radical pode ainda voltar a sofrer cisão b e recomeçar o ciclo. Um único radical pode portanto dar origem a muitas centenas de moléculas de produtos da reacção. Estas reacções denominam-se reações de propagação de radicais. Quando é que terminará a cadeia? Quando dois radicais se encontrarem:
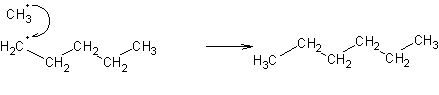
Se a concentração de radicais fôr baixa, a probabilidade de dois deles se encontrarem será muito baixa. Nessas condições, a cadeia poderá continuar uns milhares (ou dezenas de milhares) de ciclos antes de terminar.
Ao contrário dos carbocatiões, os radicais que se formam têm tendência a ser radicais primários. Porquê?
Os radicais metilo que se formam têm de reagir com hidrocarbonetos mais longos (para dar início às reacções de propagação, cisões b, etc.). Esta reacção consiste em retirar um átomo de hidrogénio da cadeia. E de onde é mais fácil retirar um hidrogénio? Das pontas da cadeia, porque é aí que existe mais espaço livre! (O carbono está rodeado de três hidrogénios que ocupam pouco espaço, e é portanto mais fácil o radical metilo aproximar-se). A outra possibilidade para produzir radicais não primários seria a isomerização dos radicais primários. Esta reação, apesar de termodinamicamente favorecida (porque os radicais secundários são mais estáveis do que os primários, e os terciários são mais estáveis do que os secundários), não é favorecida por ser mais lenta do que as reacções de propagação.
Luz visível de comprimento de onda inferior a 600 nm pode causar a cisão homolítica do peróxido de hydrogénio (HOOH) e do Br2, ao passo que a cisão do Cl2 exige fotões mais energéticos, de comprimento de onda inferior a 490 nm. Os radicais que assim se formam têm tendência a provocar a quebra (homolítica) de outras moléculas (de tipo (A-B), dando origem a um radical novo (A∙) e a uma nova ligação (entre o radical original e o resto da molécula quebrada (B∙). Se o processo total for endergónico (i.e. exigir energia) a molécula que é cindida pelo radical será normalmente aquela cuja quebra for menos dispendiosa energeticamente. Se o processo libertar energia a reacção ocorrerá imediatamente, ou seja, sem “procurar” outras possibilidades de reacção ainda mais favoráveis. Na tabela abaixo indicam-se as energias necessárias para a cisão homolítica de várias ligações.
| Ligação | Energia (kca/mol) | Ligação | Energia (kca/mol) |
| HO-OH | 47 | H-OH | 119 |
| H3C-H | 105 | H-Br | 87.4 |
| (H3C)CH2-H | 101 | H-Cl | 103 |
| (H3C)2CH-H | 98.6 | Br-Br | 46.3 |
| (H3C)3C-H | 96.5 | Cl-Cl | 58 |
| R3C-Br | 74 | R3C-Cl | 85 |
| H2C=CH-H | 111 | H2C=C(CH3)-H | 89 |
| Reacção | energia (kcal/mol) |
| H3C-H + .Br → H3C. + HBr | 105-87.4 = 17.6 |
| (CH3)H2C-H + .Br → (CH3)H2C. + HBr | 101-87.4 = 13.6 |
| (CH3)2HC-H + .Br → (CH3)2HC. + HBr | 98.6-87.4 = 10.2 |
| H3C-H + .Cl → H3C. + HCl | 105-103 = 2 |
| (CH3)H2C-H + .Cl → (CH3)H2C. + HCl | 101-103 = -2 |
| (CH3)2HC-H + .Cl → (CH3)2HC. + HCl | 98.6-103 = -4.4 |
 | Organic
Chemistry, 7th Edition, Solomons & Fryhle Um texto clássico, pormenorizado e escrito de forma muito clara. |
 | Organic
Chemistry: a Brief Course, Carey & Atkins Um bom resumo de química orgânica. |
 | Organic Chemistry, Francis Carey Bastante pormenorizado, este livro inclui um óptimo programa de construção tridimensional de moléculas, bastante útil para o estudo da estereoquímica, conformações, etc. |
"Química Orgânica", Carlos Corrêa Centro de Investigação em Química da Universidade do Porto |