Professor Associado, Universidade Fernando Pessoa
| Outras páginas de Química Orgânica: | |||
| Bioquímica metabólica: |
O átomo de carbono possui uma orbital de valência s e três orbitais de valência p.
As orbitais atómicas de dois átomos distintos podem combinar-se, de forma a formar orbitais moleculares, i.e. formarem uma ligação química. Uma orbital s pode-se combinar com outra orbital s (ou com uma orbital p) para formar uma ligação s. Duas orbitais p podem-se combinar entre si para formar uma ligação p. Só se formarão ligações p se não fôr possível formar ligações s.
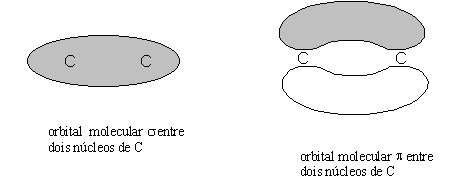
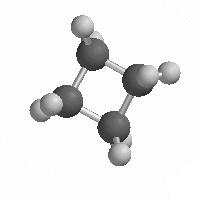
Como as três orbitais p são perpendiculares entre si, seria de esperar que as orbitais moleculares formadas com essas orbitais também fossem perpendiculares. Acontecerá isso na realidade?
Apresentam-se a seguir as estruturas de alguns compostos de carbono simples:
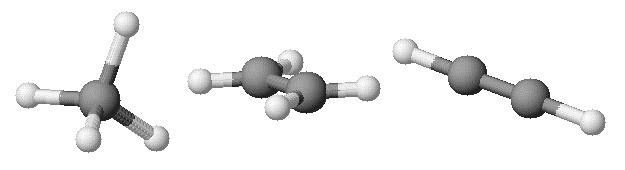
Verifica-se que na molécula de metano (tetraédrica) os ângulos de ligação são de aproximadamente 109,5 º. No etileno são de 120º e no acetileno são de 180 º. Como é que isto se pode explicar?
No caso do metano, a orbital s mistura-se com as 3 orbitais p, dando origem a quatro novas orbitais “mistas” : as orbitais híbridas sp3. Estas quatro orbitais terão energia inferior à energia das orbitais s e p que lhes deram origem, sendo por isso mais estáveis. Estas quatro orbitais, sendo todas iguais, dispôr-se-ão no espaço de forma a formar ângulos iguais entre si. Essa disposição será tetraédrica, dando origem aos ângulos observados de 109,5 º. Sempre que um átomo de carbono estiver ligado a quatro outros átomos as suas orbitais hibridizarão de forma a criar orbitais sp3.
O etileno possui uma ligação dupla. Estas ligações (tal como as ligações triplas) são formadas por orbitais moleculares p. Cada átomo de carbono utilizará uma orbital p para formar a ligação, sobrando então uma orbital s e duas orbitais p, que hibridizarão dando origem a três novas orbitais sp2. Estas orbitais afastam-se o mais possível umas das outras, dando origem ao ângulo de ligação observado de 120º.
Na molécula do acetileno, cada átomo de carbono utilizará duas orbitais p para formar ligações p. A orbital s hibridiza com a orbital p restante, dando origem a duas novas orbitais sp, que se afastam uma da outra dando origem ao ângulo de ligação de 180º.
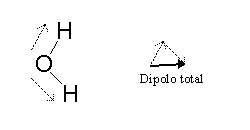
Devido à diferente electronegatividade dos átomos envolvidos numa ligação a nuvem electrónica estará deslocada em direcção do átomo mais electronegativo. O centro da carga negativa na ligação não corresponderá portanto com o centro de carga positiva (que é praticamente igual ao centro de gravidade da ligação, no caso da maioria das ligações encontradas na química orgânica). Isto dá origem a um dipolo eléctrico na ligação. O dipolo é uma grandeza vectorial, pelo que o dipolo de uma molécula será igual à soma vectorial de todos os dipolos das ligações presentes. P. ex. na molécula da água os electrões encontram-se deslocados no sentido do átomo de oxigénio. O dipolo da molécula vai ser a soma dos dipolos das duas ligações O-H.
Como sabemos, a electronegatividade aumenta ao longo de cada período da tabela periódica. Para os elementos mais comuns em química orgânica, a ordem das electronegatividades é:
H < C < N < O < F
Em cada ligação química, teremos (como dito acima) um desequilíbrio de carga electrónica na direção do átomo mais electronegativo. Estes pequenos desequilíbrios de carga denotam-se com o sinal δ (seguido de + ou - consoante existe falta ou excesso de carga electrónica). Quanto maior for o número de ligações de um átomo a átomos mais electronegativos maior será o seu δ+
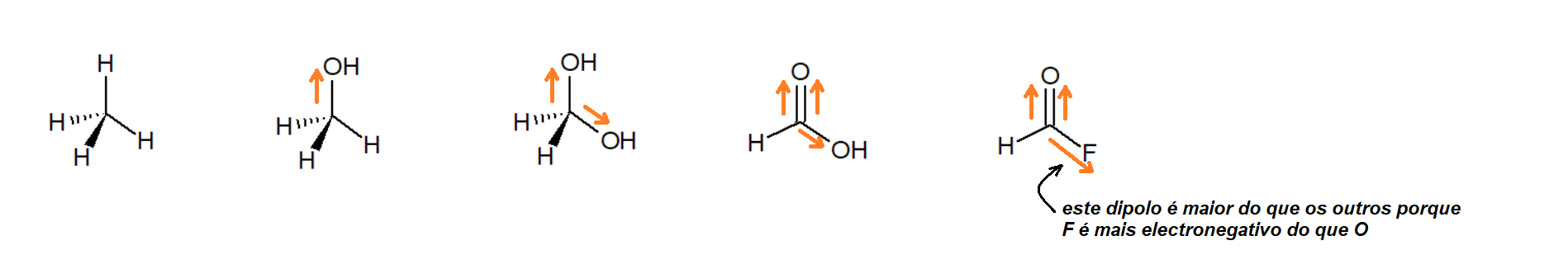
No exemplo acima a deficiência de carga electrónica no carbono central (δ+) aumenta da esquerda para a direita porque cada molécula contém mais ligações entre o carbono central e átomos electronegativos do que a molécula que está à sua esquerda.
Os hidrocarbonetos "puros" (i.e. constituídosapenas por C e H) são moléculas apolares. A introdução de átomos de elementos mais electronegativos conduz geralmente a um aumento da polaridade da molécula, desde que os dipolos não se anulem mutuamente devido à geometria e/ou simetria da molécula. Compostos polares dissolverão outros compostos polares, e compostos apolares dissolverão compostos apolares. Um composto polar não será solúvel num composto apolar, e vice-versa.
Compostos diferentes com a mesma fórmula molecular denominam-se isómeros.
Isómeros constitucionais são isómeros que diferem devido à diferente ligação dos seus átomos. Por exemplo:
| Fórmula molecular | Isómeros constitucionais |
| C4H10 | 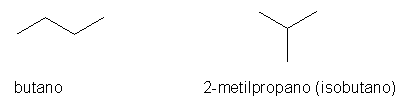 |
| C5H11Cl | 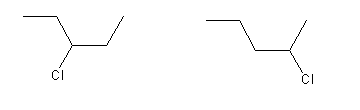 |
| C2H6O | 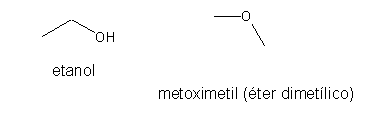 |
Esteroisómeros não são isómeros constitucionais. Os seus átomos estão ligados da mesma forma, mas diferem na sua disposição:

Estes compostos são isómeros porque não podem ser facilmente convertidos um no outro, por causa da grande barreira energética da rotação em torno da ligação dupla. Os esteroisómeros podem ser subdivididos em duas categorias: enantiómeros e diasteroisómeros.
Os enantiómeros são isómeros cujas moléculas são reflexões não sobreponíveis uma na outra:
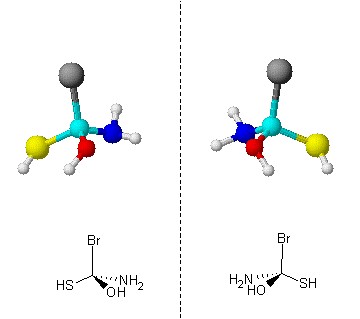
Só ocorrem enantiómeros quando a molécula é quiral. Um objecto é quiral quando não é sobreponível com a sua imagem no espelho. Por exemplo, a reflexão da mão esquerda não é sobreponível com a mão esquerda, mas com a mão direita. As mãos são, por isso, quirais.
Uma forma de reconhecer a possibilidade de existência de enantiómeros é identificar se na molécula existe um átomo tetraédrico com quatro substituintes diferentes. Trocar dois destes substituintes entre si converte um enantiómero no outro (ver exemplo acima). Esta reacção não ocorre espontaneamente, uma vez que exigiria a quebra de ligações em torno do carbono, o que exige uma energia considerável.
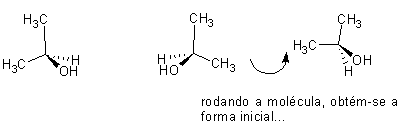
Se todos os átomos tetraédricos numa molécula têm dois substituintes iguais a molécula será aquiral (ex.: o 2-propanol)
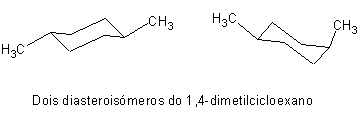
Diasteroisómeros são moléculas que não são reflexões uma da outra:
A nomenclatura dos enantiómeros e diasteroisómeros apresenta problemas particulares, que são tratados em secção própria.
As moléculas quirais provocam a rotação do plano de polarização da luz que incide nas suas soluções. Diz-se por isso que são "opticamente activas". Se uma solução contiver quantidades iguais dos dois isómeros opticamente activos de uma molécula com um único carbono quiral, a plano de polarização da luz que atravessa a solução não é alterado, porque cada fotão encontra no seu percurso exactamente tantas moléculas que lhe rodam o plano de polarização para um sentido quantas as moléculas que lhe rodam o plano de polarização para o sentido contrário. Nessas circunstâncias, diz-se que estamos em presença de uma "mistura racémica".
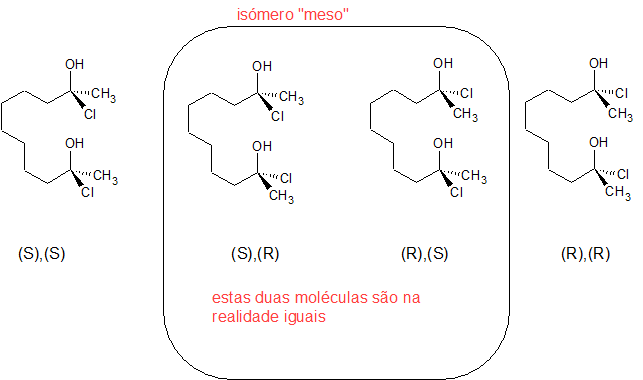
Uma molécula com n centros quirais pode por isso ter até 2n isómeros. Estes podem ser facilmente enumerados indicando a configuração (R ou S - ver a página de nomenclatura para a definição destes termos)) de cada uma deles. Existe uma excepção: quando a molécula tem dois centros quirais exactamente iguais, a configuração RS é igual à configuração SR. Curiosamente, este isómero náo é opticamente activo porque o plano de polarização da luz , ao encontrar o segundo centro quiral, será rodado exactamente na mesma intensidade que ao encontrar o primeiro centro quiral (porque os dois centros são electronicamente iguais, por terem os mesmo substituintes) mas no sentido oposto (por ter a configuração oposta). O balanço dos dois efeitos é exactamente igual a zero, e este isómero tem portanto um nome especial: chama-se isómero "meso"
Examinemos o etano:

As representações ao centro e à direita denominam-se Representações de Newman. Representam o que um observador veria se olhasse para a molécula no sentido de uma das ligações carbono-carbono. P. ex., uma representação do butano seria:
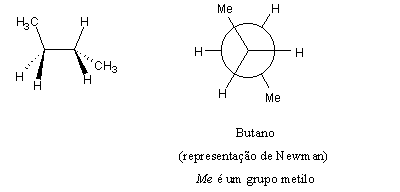
Sabemos que é possível a rotação livre em torno das ligações simples. Mas nem todas as conformações possíveis têm a mesma energia: as conformações eclipsadas são desfavoráveis, por causa das repulsões entre as nuvens electrónicas dos substituintes. As conformações mais estáveis serão as conformações alternadas. Existem dois tipos principais de conformações alternadas, segundo as posições relativas dos substituintes de cada um dos carbonos. Anti é a mais estável, porque aí é máximo o afastamento entre os substituintes.

Os anéis dos cicloalcanos maiores do que o ciclopropano não são planos: se o fossem, ocorreriam conformações eclipsadas, que são desfavoráveis.
|
|
Ciclobutano (vista de topo) |
Ciclobutano (vista de perfil) note como se evitou a conformação eclipsada |
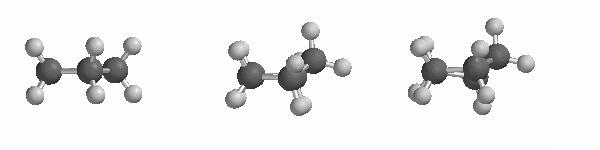
Ciclopentano (vistas de perfil). À esquerda, um hipotético ciclopentano plano. Note a conformação absolutamente eclipsada dos hidrogénios. Ao centro e à direita, duas conformações não planas, que permitem diminuir em menor ou maior grau a conformação eclipsada.
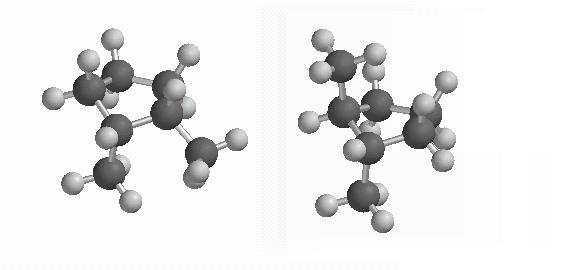
Ciclopentanos substituídos (vistas de perfil). À esquerda, um ciclopentano substituído em cis. À direita, um ciclopentano substituído em trans. Quanto maior o tamanho dos substituintes, mais favorecida será a conformação trans em relação à cis, devido a factores estéricos.
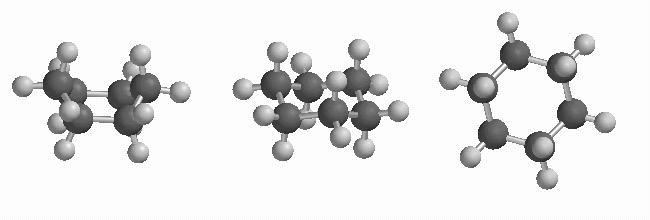
Ciclohexano (vistas de perfil). À esquerda,a conformação “em barco”. Ao centro, a conformação em “cadeira”. Note que na conformação “em barco”existem hidrogénios muito próximos. Á direita, vista de topo da conformação em “cadeira”. Cada hidrogénio axial está razoavelmente próximo de dois outros hidrogénios. Os hidrogénios equatoriais têm muito mais espaço livre à sua volta.
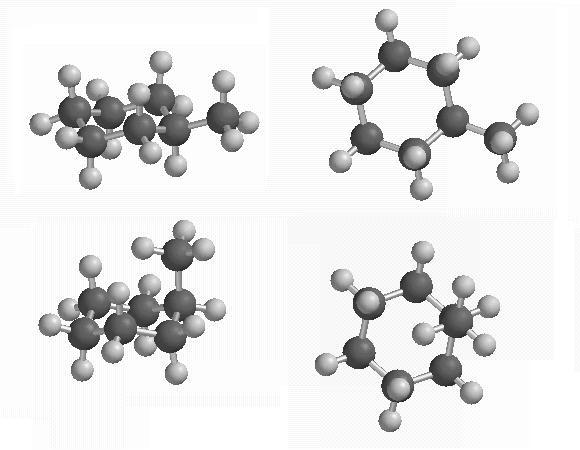
Ciclohexanos substituídos (vistas de perfil e topo). Em cima, substituição axial de um ciclohexano. Notar a pequena distância entre os hidrogénios do grupo metilo e os outros hidrogénios axiais. Em baixo, substituição equatorial do ciclohexano. A vista de topo revela que ao contrário do que poderia parecer neste caso o grupo metilo consegue estar bem afastado dos hidrogénios equatoriais. Grupos grandes preferem posições equatoriais.
 | Organic
Chemistry, 7th Edition, Solomons & Fryhle Um texto clássico, pormenorizado e escrito de forma muito clara. |
 | Organic
Chemistry: a Brief Course, Carey & Atkins Um bom resumo de química orgânica. |
 | Organic Chemistry, Francis Carey Bastante pormenorizado, este livro inclui um óptimo programa de construção tridimensional de moléculas, bastante útil para o estudo da estereoquímica, conformações, etc. |
"Química Orgânica", Carlos Corrêa Centro de Investigação em Química da Universidade do Porto |