Professor Associado,Universidade Fernando Pessoa
| Outras páginas de Química Orgânica: | |||
| Bioquímica metabólica: |
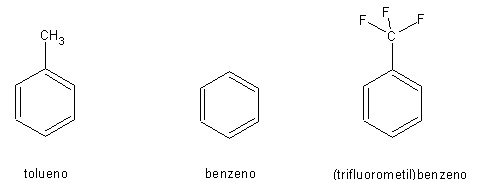
Consideremos a nitração do benzeno, do tolueno e do (trifluorometil)benzeno:
O tolueno é mais reactivo do que o benzeno. A sua nitração ocorre de 20 a 25 vezes mais depressa do que a nitração do benzeno. Ao invés, a nitração do (trifluorometil)benzeno é cerca de 40000 vezes mais lenta do que a do benzeno. O efeito é semelhante para todas as reacções de substituição electrofílica aromática. Diz-se portanto que o grupo metilo do tolueno activa o anel, e que o trifluorometilo desactiva o anel. Substituintes activantes aumentam a velocidade das reacções de substituição electrofílica aromática. Substituintes desactivantes diminuem a velocidade das reacções de substituição electrofílica aromática.
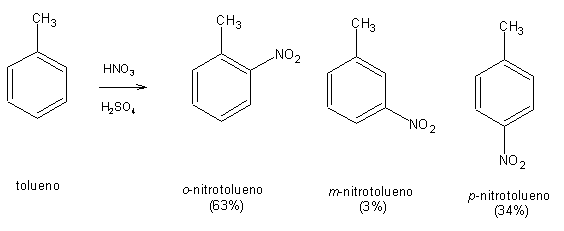
Ao contrário do benzeno, no tolueno as diferentes posições do anel não são equivalentes. A substituição no tolueno pode originar produtos substituídos em orto, meta ou para. Na nitração do tolueno, formam-se predominantemente os isómeros orto- e para-.O isómero meta- forma-se apenas em quantidades vestigiais. Diz-se por isso que o grupo metilo é orto-,para-director. A nitração do (trifluorometil)benzeno produz um resultado diferente: forma-se quase exclusivamente o isómero meta-. Diz-se portanto que o substituinte trifluorometil é um meta-director.
Já vimos que a substituição electrofílica aromática ocorre através de um intermediário: o catião cicloexadienilo. O estudo da estrutura e da estabilidade deste catião é a chave para a compreensão do efeito dos substituintes na velocidade e na regioselectividade. O princípio fundamental é: um carbocatião mais estável forma-se mais rapidamente que um carbocatião menos estável. Consideremos novamente a nitração do tolueno. O grupo metilo aumenta a densidade electrónica do anel por efeito indutivo, tornando todas as suas posições mais susceptíveis de ataque electrofílico do que as posições correspondentes no benzeno. Além disso, no caso do ataque orto, as estruturas contribuintes incluem um carbocatião terciário muito estável, devido ao efeito indutivo do grupo metilo:
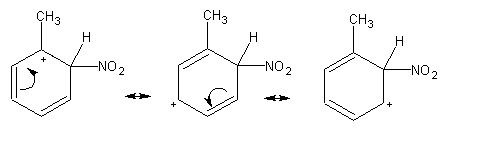
No caso do ataque meta, todas as estruturas contribuintes são carbocatiões secundários. O ataque em orto será portanto favorecido em comparação com o ataque em meta.
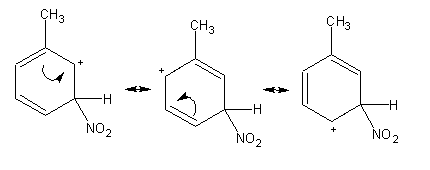
Exercício: Comprove que no caso do ataque para, também existe um carbocatião terciário entre as estruturas contribuintes.
Algo diferente se passa no caso do (trifluorometil)benzeno. O grupo trifluorometilo retira densidade electrónica ao anel por efeito indutivo, tornando todas as suas posições menos susceptíveis de ataque electrofílico do que as posições correspondentes no benzeno. Tal como no tolueno, o ataque do (trifluorometil)benzeno pelo NO2+ em posição orto dá origem a um carbocatião terciário entre as estruturas contribuintes:
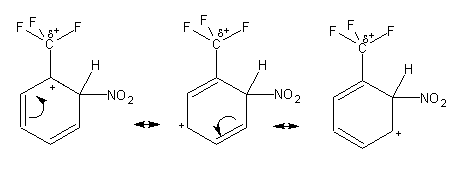
No entanto este carbocatião terciário é extremamente instável: a grande electronegatividade dos átomos de flúor cria uma deficiência electrónica significativa no carbono (assinalada por d+). O carbocatião terciário possui então duas cargas positivas em átomos adjacentes, o que o instabiliza bastante. O mesmo acontece quando o ataque é realizado em para. O ataque em meta- não tem este inconveniente, pelo que o produto da nitração do (trifluorometil)benzeno será quase exclusivamente o isómero meta-.
Exercício: Comprove que no caso do ataque meta-, não existem estruturas contribuintes em que o carbocatião esteja instabilizado.
EFEITO NA VELOCIDADE |
SUBSTITUINTE |
EFEITO NA ORIENTAÇÃO |
Muito fortemente activantes |
-NH2 ,-NHR,-NR2, -OH |
Orto-, para-directores |
Fortemente activantes |
-OR, |
|
Activantes |
-R, -CH=CR2, |
|
Desactivantes |
-F, -Cl, -Br, -I |
|
Fortemente desactivantes |
|
Meta-directores |
Muito fortemente desactivantes |
-CF3, -NO2 |
Conforme se pode ver na tabela junta, vários substituintes activantes orto-,para-directores contêm um átomo de oxigénio ou de azoto ligado ao anel benzénico. Os catiões cicloexadienilo formados por ataque orto- ou para- são estabilizados por doação de um par de electrões não compartilhados do oxigénio ou azoto. Observemos o intermediário formado por ataque orto- de um anel contendo um grupo alcóxi (-OR).
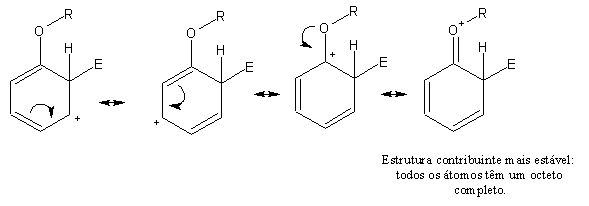
O par de electrões não compartilhados do oxigénio estabiliza o catião originado de ataque para- da mesma forma, mas o catião originado pelo ataque meta- não beneficia desta estabilização. Conforme ilustrado por este exemplo, substituintes com pares de electrões não compartilhados ligados ao anel são orto-,para-directores. Todos os substituintes activantes estabilizam o catião doando densidade electrónica ao anel. Além disso, todos os substituintes activantes são orto-,para-directores.
Todos os substituintes meta- directores são desactivantes, i.e., reagem mais lentamente do que o benzeno. Geralmente, os substituintes desactivantes encontram-se ligados ao anel benzénico através de átomos com uma carga positiva (total ou parcial), cujo efeito é retirar densidade electrónica do anel e diminuir a estabilidade do catião cicloexadienilo. Um exemplo comum é o grupo carbonilo (C=O). A ligação carbono-oxigénio é bastante polarizada, e o átomo de carbono (o menos electronegativo do par) é a zona positiva do dipolo:
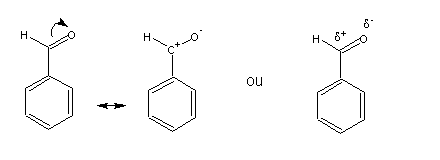
Os intermediários da nitração do benzaldeído são:
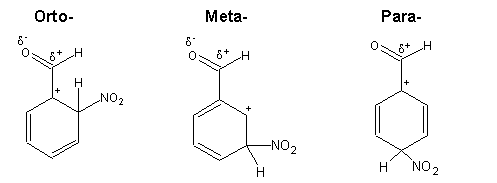
A nitração do benzaldeído dá origem quase exclusivamente ao isómero meta-, uma vez que os intermediários dos ataque em orto- e em para- são instáveis devido à presença de cargas positivas em átomos adjacentes.
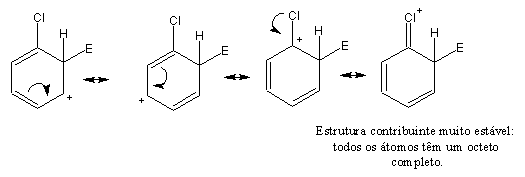
Os halogénios são substituintes peculiares: apesar de desactivantes, são orto-,para-directores. O seu efeito desactivante é devido à sua elevada electronegatividade, que provoca diminuição de densidade electrónica no anel. No entanto, tal como o oxigénio e o azoto, os halogénios possuem pares de electrões não compartilhados que podem ser doados ao carbono carregado positivamente, o que estabiliza os intermediários derivados de ataque orto- e para-:
 | Organic
Chemistry, 7th Edition, Solomons & Fryhle Um texto clássico, pormenorizado e escrito de forma muito clara. |
 | Organic
Chemistry: a Brief Course, Carey & Atkins Um bom resumo de química orgânica. |
 | Organic Chemistry, Francis Carey Bastante pormenorizado, este livro inclui um óptimo programa de construção tridimensional de moléculas, bastante útil para o estudo da estereoquímica, conformações, etc. |
"Química Orgânica", Carlos Corrêa Centro de Investigação em Química da Universidade do Porto |