Professor Associado, Universidade Fernando Pessoa
| Outras páginas de Química Orgânica: | |||
| Bioquímica metabólica: |
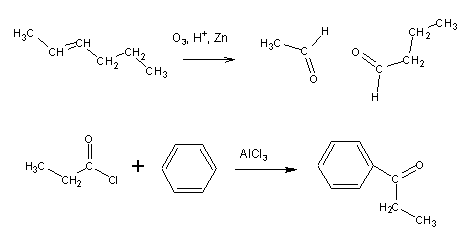
Já encontrámos várias reacções que produzem aldeídos e/ou cetonas: a oxidação dos álcoois, a ozonólise e a acilação de Friedel-Crafts:
A natureza polar do grupo carbonilo permite-lhe reagir quer com electrófilos como com nucleófilos: os nucleófilos atacarão o carbono (que tem deficiência de electrões) e os electrófilos atacarão o oxigénio (que tem elevada densidade electrónica).
Os aldeídos e as cetonas podem ser reduzidos respectivamente a álcoois primários e a álcoois secundários. A redução pode ser realizada (tal como a redução de alcenos) por hidrogenação na presença de Pt, Pd, Rh ou Ni.
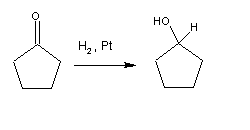
Para a maior parte das aplicações laboratoriais, este método foi substituído por métodos baseados em hidretos metálicos. Os reagentes mais comuns são o boroidreto de sódio (NaBH4) e o LiAlH4.
Os aldeídos e as cetonas reagem com a água num equilíbrio rápido:
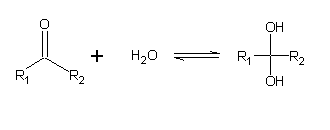
O produto é um diol (uma molécula com dois grupos álcool). A quantidade de hidrato presente no equilíbrio é muito variável, e é normalmente muito maior para os aldeídos do que para as cetonas. A posição do equilíbrio depende de factores electrónicos e de factores estéricos (relacionados com o volume). Substituintes que libertam densidade electrónica (por exemplo, grupos metilo) estabilizam o grupo carbonilo e diminuem a extensão da hidratação. Da mesma forma, substituintes que instabilizam o carbonilo favorecem a formação do hidrato. Factores estéricos: No hidrato, o carbono central está muito mais “apertado” do que no aldeído ou cetona inicial. Quanto maiores forem R1 e R2, maior será a instabilização da forma hidratada, e portanto a quantidade de hidrato presente no equilíbrio será menor. A hidratação dos aldeídos e cetonas pode ser catalizada quer por ácidos quer por bases.
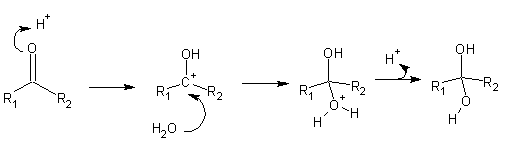
Sob condições de catálise ácida, os aldeídos reagem com álcoois formando diéteres denominados acetais. O primeiro passo é o ataque (catalizado por ácido) do aldeído por uma molécula de álcool, num processo bastante semelhante ao ocorrido na hidratação:
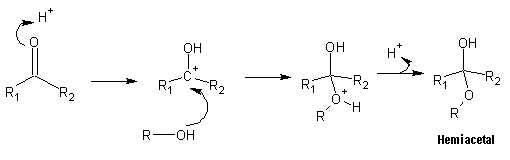
Nas condições ácidas da sua formação, o hemiacetal é convertido num carbocatião:
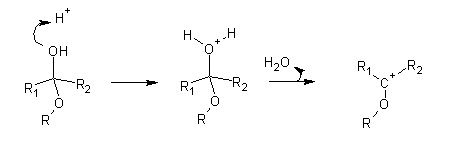
Este carbocatião é estabilizado por ressonância, devido à presença dos pares de electrões não-ligantes do oxigénio:
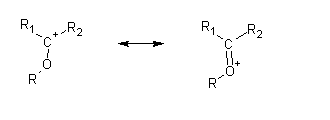
O acetal é formado por reacção do carbocatião com outra molécula de álcool:
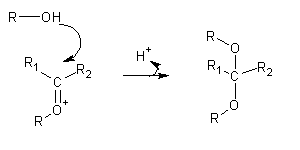
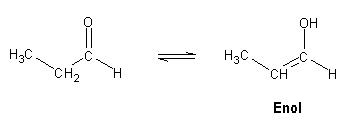
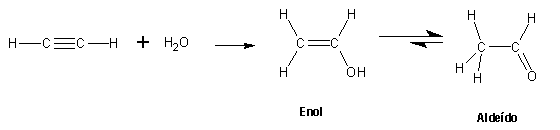
Aldeídos e cetonas com pelo menos um hidrogénio a (hidrogénio ligado ao carbono imediatamente adjacente ao carbonilo) encontram-se em equilíbrio com um isómero denominado enol. Este equilíbrio chama-se tautomerismo ceto-enol. Os tautómeros são isómeros que diferem entre si apenas na posição de um átomo ou grupo de átomos. Para cetonas e aldeídos simples, o equilíbrio encontra-se fortemente deslocado no sentido do composto carbonilo. Isto permite sintetizar aldeídos a partir de alcinos: a hidratação de um alcino dá origem a um enol, que está em equilíbrio com o composto carbonilo correspondente:
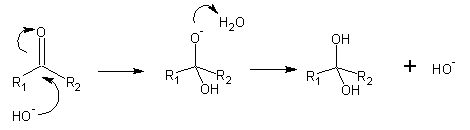
A enolização pode ser catalizada por uma base forte, como o anião hidróxido. A base retira um hidrogénio a, dando origem ao enolato correspondente. O enolato é uma espécie estabilizada por ressonância:
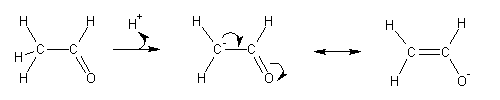
Esta ressonância é a razão da elevada acidez dos hidrogénios a dos aldeídos e cetonas, uma vez que o anião enolato é mais estabilizado do que o composto carbonilo original.
O aldeído pode ser parcialmente convertido em enolato por catálise alcalina. O enolato assim formado pode atacar outra molécula de aldeído dando origem a um aldol (uma molécula com um grupo aldeído e um grupo álcool). Apresenta-se a seguir o mecanismo desta reacção (adição aldólica)
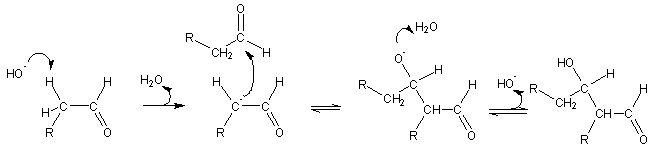
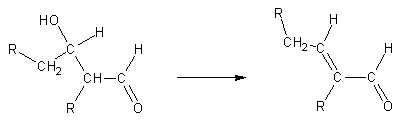
A adição aldólica é um equilíbrio que favorece os produtos quando o reagente é um aldeído. Quando se usam cetonas o equilíbrio normalmente encontra-se deslocado no sentido dos reagentes; as cetonas dão por isso fracos rendimentos de produtos de adição aldólica. O produto da adição aldólica de um aldeído é um aldeído b-hidroxilado. Da mesma forma, sob determinadas condições, um aldeído (ou cetona) b-hidroxilado pode ser quebrado em duas moléculas mais pequenas numa reacção que é o inverso da adição aldólica. Uma reacção deste tipo é a quebra de frutose-1,6-bisfosfato em dihidroxiacetona fosfatada e gliceraldeído-3-fosfato, que ocorre na glicólise . Por aquecimento, estes compostos desidratam, dando origem a uma ligação dupla carbono-carbono conjugada com o aldeído. A desidratação ocorre sempre na direcção em que se formam ligações conjugadas C=C-C=O.
 | Organic
Chemistry, 7th Edition, Solomons & Fryhle Um texto clássico, pormenorizado e escrito de forma muito clara. |
 | Organic
Chemistry: a Brief Course, Carey & Atkins Um bom resumo de química orgânica. |
 | Organic Chemistry, Francis Carey Bastante pormenorizado, este livro inclui um óptimo programa de construção tridimensional de moléculas, bastante útil para o estudo da estereoquímica, conformações, etc. |
"Química Orgânica", Carlos Corrêa Centro de Investigação em Química da Universidade do Porto |